



その他神経難病について
当クリニックには、 筋萎縮性側索硬化症(ALS)や多発性硬化症(MS)、脊髄小脳変性症(SCD)、ハンチントン舞踏病(HC)、多発性筋炎・皮膚筋炎(PM/DM)、重症筋無力症(MG)、ミオパチー、クロイツフェルト・ヤコプ病(CJD)など数多くの神経難病の患者様が通院され、専門的な治療を受けています。 お気軽にご相談ください。
筋萎縮性側索硬化症(ALS)について
について")
ALSは、英語名(Amyotrophic Lateral Sclerosis)の頭文字をとった略称です。ALSは運動神経が冒されて筋肉が萎縮していく進行性の神経難病です。
手足や顔など自分の思いどおりにからだを動かすときに必要な筋肉を随意筋と言います。この随意筋を支配する神経を運動ニューロンといいます。運動ニューロンは、歩く、物を持ち上げる、飲み込むなど、様々な動作をするときに、脳の命令を筋肉に伝える役目をしています。この運動ニューロンが侵されると、筋肉を動かそうとする信号が伝わらなくなり、筋力が低下し、筋肉が萎縮します。ALSはこの運動ニューロンが侵される病気です。
病気が進むにしたがって、手や足をはじめ体の自由がきかなくなり、次第に話すことも食べることも、呼吸することさえも困難になってくることがあります。 しかし、感覚、自律神経と頭脳が悪くなることはほとんどありません。
進行は個人差がありますが、発病して3~5年で寝たきりになり、人工呼吸器を装着しなければ呼吸することができなくなることもあります。
最初、手足の運動障害で気づかれる場合が多いのですが、「ろれつが回りにくい」「食べ物を飲み込みにくい」などの球麻痺症状が最初に出現する場合もあります。 最初に症状が出て数ヵ月たつと、「手足の運動障害」「会話・嚥下障害」「呼吸障害」があらわれ、その進み具合は患者さんによって異なります。これらの症状の進行に応じて担当医に相談し、さまざまな対処法をとる必要があります。
当クリニックではケアマネージャーや訪問看護師、理学・作業・言語療法士、ヘルパーらと協力し、少しでも気持ちよく生活できるよう支援しています。
ALSの症状と障害部位
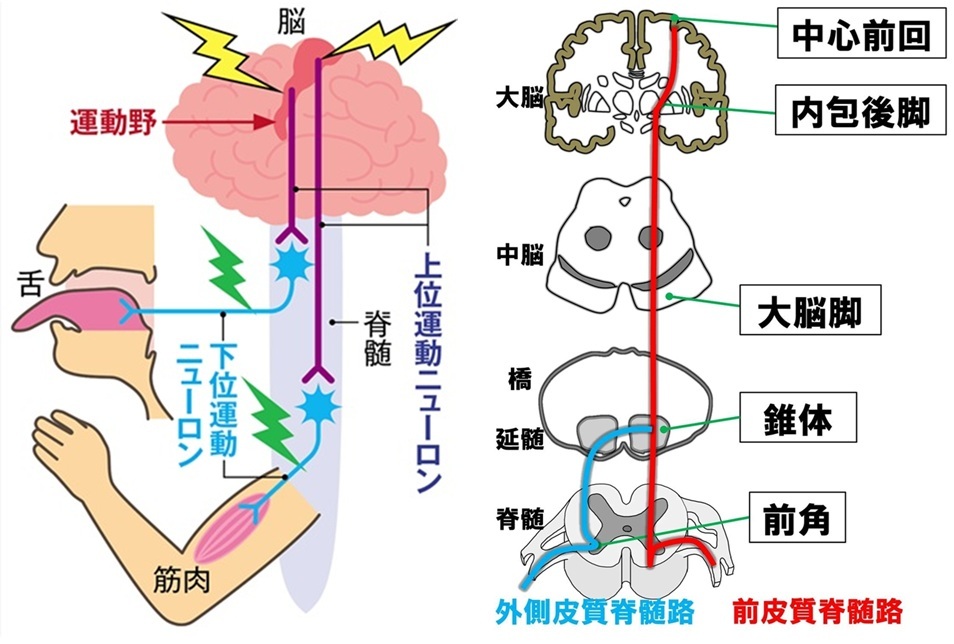
運動ニューロン
・下位運動ニューロン:
脊髄前角細胞で,骨格筋に随意運動のための刺激を送る
・上位運動ニューロン:
大脳皮質の運動神経細胞で,脊髄前角細胞に随意運動のための刺激を送る
◎運動ニューロン病
運動神経細胞がゆっくりと変性
・上・下位ともに障害→筋萎縮性側索硬化症(ALS)
・下位だけ障害→脊髄性筋萎縮性
・上位だけ障害→原発性側索硬化症
症状は筋萎縮と筋力低下が中心
→四肢筋力低下と筋萎縮,構音障害,嚥下障害,呼吸障害など
感覚障害や排尿障害,眼球運動障害が出現することは少ない
※人工呼吸器による長期生存例では出現することあり
ALSの発症機序
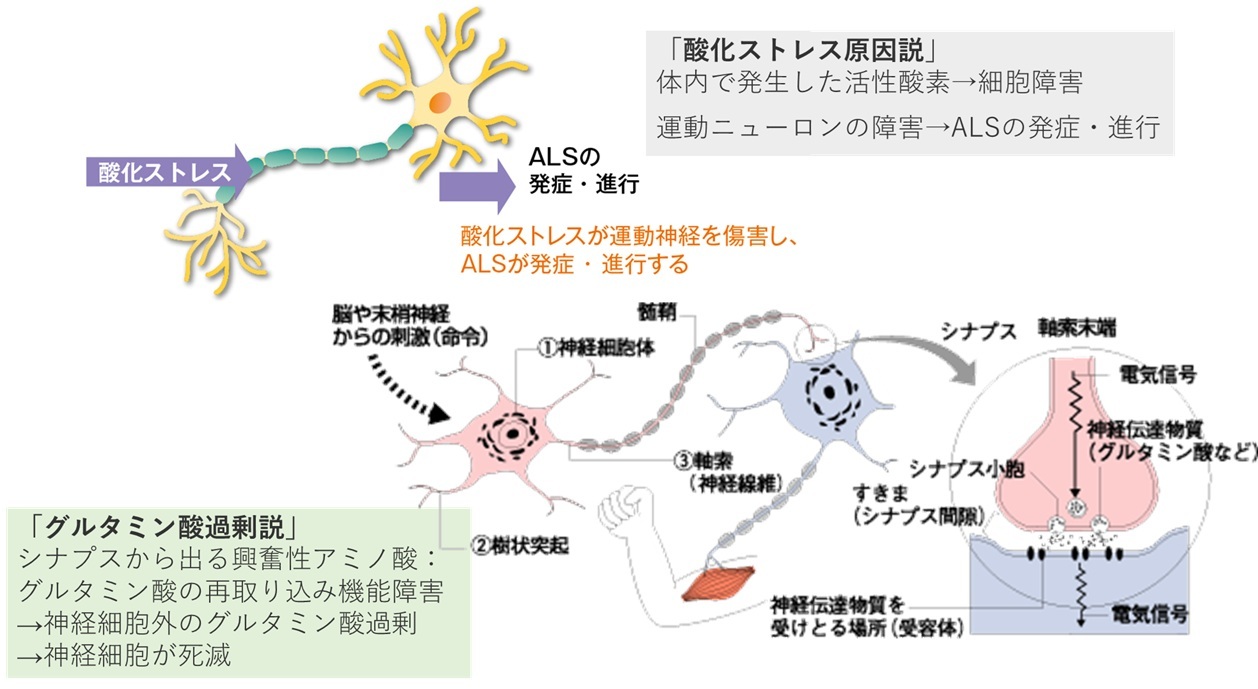
「グルタミン酸過剰説」
シナプスから出る興奮性アミノ酸:グルタミン酸の再取り込み機能障害
→神経細胞外のグルタミン酸過剰
→神経細胞が死滅
「酸化ストレス原因説」
体内で発生した活性酸素→細胞障害
運動ニューロンの障害→ALSの発症・進行
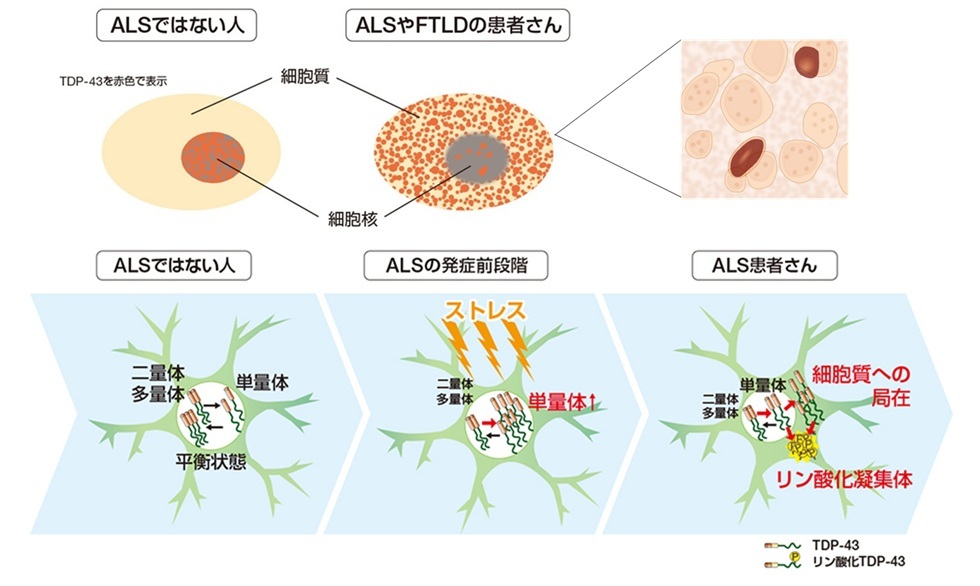
ALSの病因となるTDP-43(TAR DNA-binding protein 43)
ALS,FTD患者の運動神経細胞に「ユビキチン陽性封入体」
ユビキチン陽性封入体=TDP-43が凝集したもの
TDP-43は細胞核に存在するRNA結合蛋白で,細胞の蛋白合成の制御に関与
ALS,FTDでは細胞質にも存在
◎細胞核から細胞質に移行する仕組みの解明→ALSやFTDの治療につながる可能性
ALS治療薬(2025年現在)
リルゾール
・ALS発症の仮説:グルタミン酸過剰説
グルタミン酸が神経細胞のシナプス間隙に過剰に存在
→神経細胞が常に興奮状態=早期に死滅
◎グルタミン酸による興奮毒性を抑制→神経保護作用
・生存期間を平均2〜3
ヵ月延長する延命効果
・運動機能や筋力に対する改善効果は期待し難い
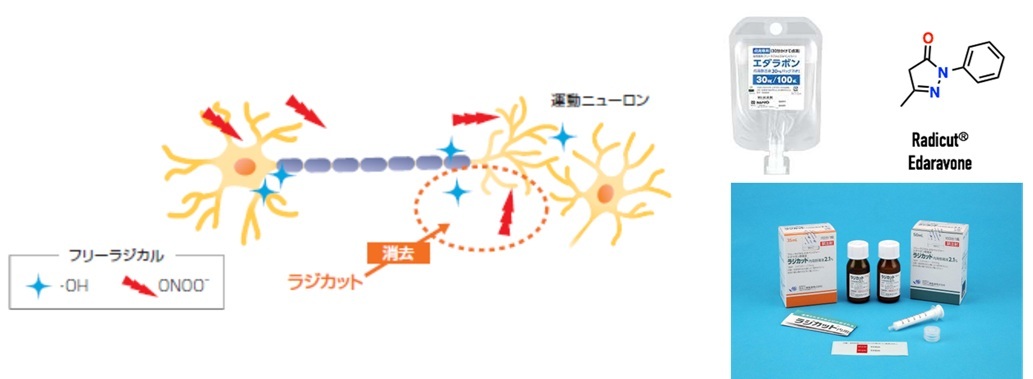
エダラボン
・ALS発症の仮説:酸化ストレス原因説
フリーラジカルを消去→運動ニューロンの障害抑制
・軽度ALSにエダラボンを6か月間投与
→ALS機能評価スケール(ALSFRS-R)の日常生活動作障害進行を約33%抑制
・ラジカット内用懸濁液=ラジカット注30mgと同一有効成分
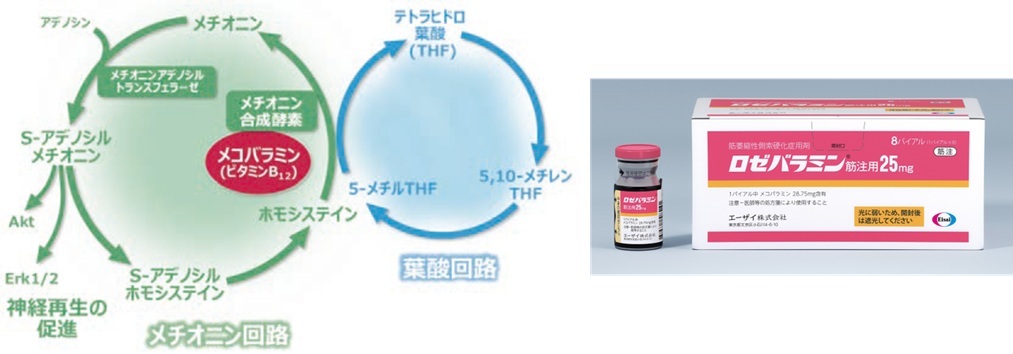
メコバラミン
・メコバラミンのALSの病態に対する作用機序は不明
・神経保護作用,神経軸索再生作用により有効の可能性
・軽度ALSにロゼバラミンを16周間投与
→ALS機能評価スケール(ALSFRS-R)の日常生活動作障害進行を約43%抑制
・投与方法:ロゼバラミン50mgを1日1回50mg,週2回筋注
脊髄小脳変性症(SCD)について
について")
SCDは英語名(SpinoCerebellar Degeneration)の頭文字をとった略称です。
脊髄と小脳の神経細胞が徐々に減少し、ふらつき(運動失調)が増強していく進行性の神経難病で、多くの種類があります。発現様式により遺伝性と非遺伝性とに分けられます。
非遺伝性は、脊髄小脳変性症の50-60%を占め、多系統萎縮症や晩発性小脳皮質萎縮症などが含まれます。
遺伝性は、脊髄小脳変性症の40-50%を占めます。この中で、遺伝子型がわかっているものが約8割、遺伝子型が未定のものが約2割です。遺伝子型がわかっているものの中には、脊髄小脳性失調症1(SCA1), 脊髄小脳性失調症2(SCA2), 脊髄小脳性失調症3(SCA3, マチャド・ジョセフ病), 脊髄小脳性失調症6(SCA6), 歯状核赤核淡蒼球ルイ体萎縮症(DRPLA)などがあります。
遺伝性SCDの原因遺伝子の一部にCAGまたはGAA反復(トリプレットリピート)の異常延長があることが明らかとなり、機能障害との関連が注目されています。本邦では、SCA3、SCA6が多くみられます。
小脳性の運動失調には内服薬(セレジスト)や注射薬(ヒルトニン)が有効なこともあります。